| Chapter 14. COSMO Solvation Model | ||
|---|---|---|

|
Part I. AMPAC™ 10 User Guide |  |
| Chapter 14. COSMO Solvation Model | ||
|---|---|---|
|
|
Part I. AMPAC™ 10 User Guide | |
Table of Contents
COSMO-RS is the solvation model of Andreas Klamt. COSMO is an abbreviation for “Conductor-like Screening Model” and belongs to the class of dielectric continuum models.[39] COSMO-RS is an extension of the original COSMO model, where the RS stands for “Real Solvents.”[40]
When COSMO is used, parameters for water are assumed. These parameters may be overridden by the use of keywords. The solvent’s dielectric constant can be set by DIELEC (or EPS). Similarly, the index of refraction of the solvent can be set by IOFR (or REFRACT). Similarly the solvent’s molecular radius for use in the integration is specified by RSOLV.
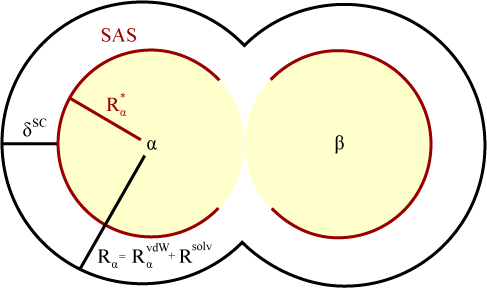
The molecular surface is defined in COSMO as the sum of overlapping van der Waals radii, RαvdW, about each atom, α. (These van der Waals radii can be set by keyword VDW.) The solvent is approximated as a sphere of radius, Rsolv (which is set by RSOLV). The surface available to the solvent’s centers is therefore given as the surface defined by the sum of overlapping radii, Rα, where Rα = RαvdW + Rsolv. The effective charges which are responsible for the dielectric screening will not be located at the centers of solvent molecules but instead located at distance, δSC from the molecular center. (The effective molecular radius, δSC, is set by DELSC.) The solvent accessible surface is then defined by the sum of overlapping radii, Rα*, where Rα* = Rα - δSC. (By default, DELSC is set to being equal to RSOLV, which makes Rα* = RαvdW. In general, DELSC should not be adjusted.)
The basic grid for the solvent accessible surface is constructed for each individual atom, α, with points distributed evenly on a sphere of radius Rα. Grid points for atom, α, that fall within the van der Waals radii of another atom, β, will be discarded so that each grid point is uniquely associated with a single atom. Surviving points are then contracted to the sphere of radius, Rα*, which then represents the allowed positions for the screening charges and define the solvent accessible surface. Each of the screening points are grouped into compact segments. NSPA (number of segments per atom) determines the number of segments used and may be specified by the user. (Increasing NSPA improves accuracy but also correspondingly increases computation time.) This algorithm the procedure to generate a relatively small number of segments composed of sets of basic points needed to compute the segment interaction components.
The electrostatic interaction energies are computed between distinct pairs of solvation segments. The interaction between two segments is calculated simply as the sum of pairwise Coulomb interactions over the points in each segment. If the distance between segments is greater than a certain distance (set by DISEX), then a two point approximation is used in place of an exact summation.
COSMO has been updated to use the 1998 parameters of Klamt, et. al.[41] To use the original 1993 parameters for backwards compatability with older versions of AMPAC, use the COSMO=1993 keyword.
This section contains an alphabetical list of all COSMO keywords. Note, certain COSMO dedicated keywords, are also used with AMSOL’s SM5C and SM5CR models.
|
Invoke the COSMO solvation model. |
|
|
Write out data for further COSMO processing. |
|
|
Specify the effective molecular radius of the desired solvent. |
|
|
Specify the dielectric constant for desired solvent. (Equivalent to EPS) |
|
|
Distance threshold for using two-point interaction approximation. |
|
|
Specify the dielectric constant for desired solvent. (Equivalent to DIELEC) |
|
|
Specify the index of refraction of the desired solvent. (Equivalent to REFRACT) |
|
|
Specify the number of segments per atom. |
|
|
Use old MINDO3 parameters with COSMO. |
|
|
Specify the index of refraction of the desired solvent. (Equivalent to IOFR) |
|
|
Specify the molecular radius of the desired solvent. |
|
|
Specify an element’s van der Waals radius. |
|
Copyright © 1992-2013 Semichem, Inc. All rights reserved. |
|
|
 |
|
| Chapter 13. Simulated Annealing |  |
Chapter 15. AMSOL Model Module |