| Chapter 26. Two-Dimensional Grid Search | ||
|---|---|---|

|
Part II. AMPAC™ 10 Examples |  |
| Chapter 26. Two-Dimensional Grid Search | ||
|---|---|---|
|
|
Part II. AMPAC™ 10 Examples | |
Table of Contents
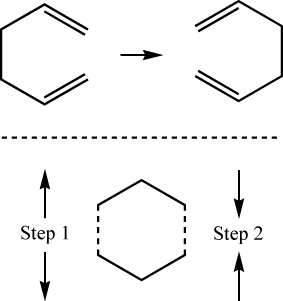
A two-dimensional grid search calculates the properties of geometries corresponding
to a set of points in a grid of user-defined size in two dimensions. In many ways, it is
analogous to the reaction coordinate method described above, although two variables are changed
and not as much information is provided. The final result is an 11×11 matrix of energies (121 in
all) corresponding to sites on the grid. A grid search is called for if the keywords
STEP1=n.n and
STEP2=m.m are on the
keyword line and two coordinates are marked with “-1” as the optimization flags.
They are related to STEP1 and STEP2 in order of their definition. The reaction described in this
file is part of the surface describing the inversion of 1,5-hexadiene:
am1 rhf singlet qcscf t=auto step1=0.15 step2=0.15 grad vis=min1,5-HEXADIENE INVERSION Rxn grid calculation C 0.000000 0 0.000000 0 0.000000 0 0 0 0 C 1.300000 1 0.000000 0 0.000000 0 1 0 0 C 1.540000 1 130.000000 1 0.000000 0 2 1 0 C 1.540000 -1 120.000000 1 0.000000 1 3 2 1
C 1.540000 1 120.000000 1 0.000000 1 4 3 2 C 2.190000 -1 110.000000 1 0.000000 1 1 2 3
H 1.000000 1 120.000000 1 90.000000 1 1 2 3 H 1.000000 1 120.000000 1 180.000000 1 1 2 7 H 1.000000 1 120.000000 1 -180.000000 1 2 3 4 H 1.000000 1 109.500000 1 120.000000 1 3 4 5 H 1.000000 1 109.500000 1 -120.000000 1 3 4 5 H 1.000000 1 109.500000 1 120.000000 1 4 3 2 H 1.000000 1 109.500000 1 -120.000000 1 4 3 2 H 1.000000 1 120.000000 1 120.000000 1 5 4 3 H 1.000000 1 120.000000 1 90.000000 1 6 5 4 H 1.000000 1 120.000000 1 180.000000 1 6 5 15 0 0.000000 0 0.000000 0 0.000000 0 0 0 0
|
A reaction grid calculation will be performed if exactly two of optimization flags are marked with a “-1”. The keywords STEP1 and STEP2 are also required for a reaction grid. By default, TRUSTE will be use to perform the constrained optimization at each of the grid points but this default can be overridden by BFGS, DFP, or EF. |
|
|
This is the first reaction coordinate and will be incremented and decremented by the value of STEP1. |
|
|
This is the second reaction coordinate and will be incremented and decremented by the value of STEP2. These two items will be used to construct a grid of points and optimizations will be carried out at each point on the surface. The point given in the geometry definition is the center of the grid and five steps are taken in each direction from this center point. The following list shows the values that the first and second reaction coordinates will take on. The center point is indicated by ×:
Step 1
0.79 0.94 1.09 1.24 1.39 1.54 1.69 1.84 1.99 2.14 2.29
1.44 o o o o o o o o o o o
1.59 o o o o o o o o o o o
1.74 o o o o o o o o o o o
S 1.89 o o o o o o o o o o o
t 2.04 o o o o o o o o o o o
e 2.19 o o o o o × o o o o o
p 2.34 o o o o o o o o o o o
2.49 o o o o o o o o o o o
2 2.64 o o o o o o o o o o o
2.79 o o o o o o o o o o o
2.94 o o o o o o o o o o o
|
This file is abbreviated to conserve space. The archive file contains the geometries corresponding to each grid point (in the order they are computed) and so contains 121 geometries. Only the first and last geometry are shown here separated by “=========” to indicate the missing geometry. A complete copy of the file is found in the test suite results directory.
Timestamp: 2011-08-31-12-50-56-0000000C90-win64
User Info: John Millam, Nahum,
SUMMARY OF AM1 CALCULATION
Aug-31-2011
AMPAC Version 10.0.1
Presented by:
Semichem, Inc.
www.semichem.com
FORMULA: C6H10
1,5-HEXADIENE INVERSION
Rxn grid calculation
GEOMETRY OPTIMIZED : ENERGY MINIMIZED
SCF FIELD WAS ACHIEVED
FINAL HEAT OF FORMATION = 35.205941 kcal
= 147.336862 kJ
ELECTRONIC ENERGY = -3921.641508 eV
CORE-CORE REPULSION = 3017.253917 eV
TOTAL ENERGY = -904.387591 eV
GRADIENT NORM = 0.122819
RMS GRADIENT NORM = 0.019419
UNSTABLE MODE(S) = 0 ( ESTIMATE )
IONIZATION POTENTIAL = 8.815998 eV
HOMO-LUMO GAP = 9.724583 eV
DIPOLE = 0.117158 debyes
MOLECULAR WEIGHT = 82.145000
MOLECULAR POINT GROUP = C2 0.100000
NO. OF FILLED LEVELS = 17 (OCC = 2)
TOTAL NUMBER OF ORBITALS = 34
COMPUTATION TIME = 0.27 SECONDS
FINAL GEOMETRY OBTAINED CHARGE
AM1 RHF SINGLET QCSCF T=AUTO STEP1=0.15 STEP2=0.15 GRAD VIS=MIN
1,5-HEXADIENE INVERSION
Rxn grid calculation
C 0.000000 0 0.000000 0 0.000000 0 0 0 0 -0.1854
C 1.344400 1 0.000000 0 0.000000 0 1 0 0 -0.1969
C 1.478626 1 121.657458 1 0.000000 0 2 1 0 -0.1251
C 1.540000 -1 109.342887 1 -68.211198 1 3 2 1 -0.1251
C 1.478615 1 109.347870 1 48.202109 1 4 3 2 -0.1969
C 2.190000 -1 95.346255 1 65.274865 1 1 2 3 -0.1854
H 1.099450 1 121.397362 1 172.236877 1 1 2 3 0.1054
H 1.100089 1 123.036405 1 166.967481 1 1 2 7 0.1136
H 1.100298 1 116.408797 1 -257.823799 1 2 3 4 0.1190
H 1.119802 1 109.026919 1 169.882036 1 3 4 5 0.0840
H 1.121496 1 108.702378 1 -72.942347 1 3 4 5 0.0854
H 1.119809 1 109.028684 1 169.886372 1 4 3 2 0.0840
H 1.121539 1 108.700345 1 -72.944721 1 4 3 2 0.0854
H 1.100291 1 116.407651 1 102.183253 1 5 4 3 0.1190
H 1.099413 1 121.399986 1 172.213054 1 6 5 4 0.1054
H 1.100088 1 123.032198 1 166.974748 1 6 5 15 0.1136
0 0.000000 0 0.000000 0 0.000000 0 0 0 0
========
Timestamp: 2011-08-31-12-50-56-0000000C90-win64
User Info: John Millam, Nahum,
SUMMARY OF AM1 CALCULATION
Aug-31-2011
AMPAC Version 10.0.1
Presented by:
Semichem, Inc.
www.semichem.com
FORMULA: C6H10
1,5-HEXADIENE INVERSION
Rxn grid calculation
GEOMETRY OPTIMIZED : ENERGY MINIMIZED
SCF FIELD WAS ACHIEVED
FINAL HEAT OF FORMATION = 155.830031 kcal
= 652.148681 kJ
ELECTRONIC ENERGY = -3850.295129 eV
CORE-CORE REPULSION = 2951.138191 eV
TOTAL ENERGY = -899.156938 eV
GRADIENT NORM = 0.128399
RMS GRADIENT NORM = 0.020302
UNSTABLE MODE(S) = 0 ( ESTIMATE )
IONIZATION POTENTIAL = 7.700880 eV
HOMO-LUMO GAP = 7.362815 eV
DIPOLE = 1.221778 debyes
MOLECULAR WEIGHT = 82.145000
MOLECULAR POINT GROUP = C2 0.100000
NO. OF FILLED LEVELS = 17 (OCC = 2)
TOTAL NUMBER OF ORBITALS = 34
COMPUTATION TIME = 22.82 SECONDS
FINAL GEOMETRY OBTAINED CHARGE
AM1 RHF SINGLET QCSCF T=AUTO STEP1=0.15 STEP2=0.15 GRAD VIS=MIN
1,5-HEXADIENE INVERSION
Rxn grid calculation
C 0.000000 0 0.000000 0 0.000000 0 0 0 0 -0.1216
C 1.527610 1 0.000000 0 0.000000 0 1 0 0 -0.2061
C 1.451551 1 112.539779 1 0.000000 0 2 1 0 -0.0841
C 2.290000 -1 108.081419 1 9.084981 1 3 2 1 -0.0838
C 1.451318 1 108.125718 1 -37.957815 1 4 3 2 -0.2062
C 1.440000 -1 114.532117 1 44.966909 1 1 2 3 -0.1218
H 1.127514 1 106.973848 1 167.723201 1 1 2 3 0.0710
H 1.127241 1 107.509394 1 113.778478 1 1 2 7 0.0903
H 1.131027 1 105.150830 1 -232.185604 1 2 3 4 0.1217
H 1.093519 1 110.753062 1 189.599419 1 3 4 5 0.0657
H 2.176064 1 117.668474 1 -66.666173 1 3 4 5 0.0624
H 1.093468 1 110.975969 1 189.351914 1 4 3 2 0.0660
H 2.175806 1 117.686710 1 -66.687426 1 4 3 2 0.0628
H 1.131067 1 105.138280 1 127.866330 1 5 4 3 0.1220
H 1.127500 1 106.978779 1 167.656614 1 6 5 4 0.0710
H 1.127231 1 107.510904 1 113.782092 1 6 5 15 0.0905
0 0.000000 0 0.000000 0 0.000000 0 0 0 0
Timestamp: 2011-08-31-12-50-56-0000000C90-win64
User Info: John Millam, Nahum,
*******************************************************************************
AM1 CALCULATION RESULTS
*******************************************************************************
* AMPAC Version 10.0.1
* Presented by:
*
* Semichem, Inc.
* www.semichem.com
*
* AM1 - THE AM1 HAMILTONIAN TO BE USED
* RHF - RESTRICTED HARTREE-FOCK CALCULATION
* STEP1 - STEP SIZE IN FIRST DIMENSION OF 2-D GRID CALCULATION
* STEP2 - STEP SIZE IN SECOND DIMENSION OF 2-D GRID CALCULATION
* VIS=MIN - AVOID STORING SURFACE AND FREQUENCY DATA IN VIS FILE
* T=AUTO - AUTOMATIC DETERMINATION OF ALLOWED TIME
* GRADIENTS- ALL GRADIENTS TO BE PRINTED
* SINGLET - IS THE REQUIRED SPIN MULTIPLICITY
* QCSCF - USE QUADRATICALLY CONVERGENT SCF METHOD
*******************************************************************************
AM1 RHF SINGLET QCSCF T=AUTO STEP1=0.15 STEP2=0.15 GRAD VIS=MIN
1,5-HEXADIENE INVERSION
Rxn grid calculation
ATOM CHEMICAL BOND LENGTH BOND ANGLE TWIST ANGLE
NUMBER SYMBOL (ANGSTROMS) (DEGREES) (DEGREES)
(I) NA:I NB:NA:I NC:NB:NA:I NA NB NC
1 C
2 C 1.30000 * 1
3 C 1.54000 * 130.00000 * 2 1
4 C 1.54000 120.00000 * 0.00000 * 3 2 1
5 C 1.54000 * 120.00000 * 0.00000 * 4 3 2
6 C 2.19000 110.00000 * 0.00000 * 1 2 3
7 H 1.00000 * 120.00000 * 90.00000 * 1 2 3
8 H 1.00000 * 120.00000 * 180.00000 * 1 2 7
9 H 1.00000 * 120.00000 * -180.00000 * 2 3 4
10 H 1.00000 * 109.50000 * 120.00000 * 3 4 5
11 H 1.00000 * 109.50000 * -120.00000 * 3 4 5
12 H 1.00000 * 109.50000 * 120.00000 * 4 3 2
13 H 1.00000 * 109.50000 * -120.00000 * 4 3 2
14 H 1.00000 * 120.00000 * 120.00000 * 5 4 3
15 H 1.00000 * 120.00000 * 90.00000 * 6 5 4
16 H 1.00000 * 120.00000 * 180.00000 * 6 5 15
MOLECULAR POINT GROUP SYMMETRY CRITERIA
C1 0.10000000
SINGLET STATE CALCULATION
RHF CALCULATION, NO. OF DOUBLY OCCUPIED LEVELS = 17
** REFERENCES TO PARAMETERS **
H (AM1): M.J.S. DEWAR ET AL, J. AM. CHEM. SOC. 107 3902-3909 (1985).
C (AM1): M.J.S. DEWAR ET AL, J. AM. CHEM. SOC. 107 3902-3909 (1985).
CARTESIAN COORDINATES
ATOM X Y Z
1 C 0.00000000 0.00000000 0.00000000
2 C 1.30000000 0.00000000 0.00000000
3 C 2.28989292 1.17970844 0.00000000
4 C 1.76318190 2.62683508 0.00000000
5 C 0.24657796 2.89425327 0.00000000
6 C -0.74902411 2.05792684 0.00000000
7 H -0.50000000 0.00000000 -0.86602540
8 H -0.50000000 0.00000000 0.86602540
9 H 1.64202014 -0.93969262 0.00000000
10 H 2.84695822 1.02723379 -0.81635148
11 H 2.84695822 1.02723379 0.81635148
12 H 2.09190985 3.10171211 0.81635148
13 H 2.09190985 3.10171211 -0.81635148
14 H -0.17063405 3.40751163 0.75000000
15 H -1.13187263 1.73632613 0.86602540
16 H -1.13187263 1.73632613 -0.86602540
"GRID" PROCEDURE TO BUILD A 2-D SURFACE FROM A 42-D HYPERSURFACE.
(VERSION 2, NOVEMBER 94).
ATTENTION: SUCH A METHOD CAN YIELD SPURIOUS DISCONTINUITIES AS OPTIMIZED
VARIABLES MAY RESPOND DISCONTINUOUSLY TO SMOOTH CHANGES IN THE DRIVING COORDS.
THE PRESENT ALGORITHM IS TUNED TO INCREASE CHANCES OF GETTING A RELIABLE
RESPONSE IN -AT LEAST- THE CENTRAL REGION OF THE GRID.
STANDARD DEVIATION ON ENERGY (KCAL) 0.00000055521
STANDARD DEVIATION ON GRADIENT (KCAL/A,RD,RD) 0.00010624 0.00020591 0.00019285
CENTRAL POINT IS LABELED (0,0)
LABELS 1ST COORD 2ND COORD HEAT OF FORM. RMS-GRAD TIME(sec)
( 0, 0) 1.540 2.190 35.205941 0.019 0.55
OPTIMIZED GEOMETRY OF CENTRAL POINT FOLLOWS.
IT DEFINES THE "GERM" FOR SUBSEQUENT OPTIMIZATIONS.
ATOM CHEMICAL BOND LENGTH BOND ANGLE TWIST ANGLE
NUMBER SYMBOL (ANGSTROMS) (DEGREES) (DEGREES)
(I) NA:I NB:NA:I NC:NB:NA:I NA NB NC
1 C
2 C 1.34440 * 1
3 C 1.47863 * 121.65746 * 2 1
4 C 1.54000 109.34289 * -68.21120 * 3 2 1
5 C 1.47861 * 109.34787 * 48.20211 * 4 3 2
6 C 2.19000 95.34626 * 65.27486 * 1 2 3
7 H 1.09945 * 121.39736 * 172.23688 * 1 2 3
8 H 1.10009 * 123.03641 * 166.96748 * 1 2 7
9 H 1.10030 * 116.40880 * -257.82380 * 2 3 4
10 H 1.11980 * 109.02692 * 169.88204 * 3 4 5
11 H 1.12150 * 108.70238 * -72.94235 * 3 4 5
12 H 1.11981 * 109.02868 * 169.88637 * 4 3 2
13 H 1.12154 * 108.70035 * -72.94472 * 4 3 2
14 H 1.10029 * 116.40765 * 102.18325 * 5 4 3
15 H 1.09941 * 121.39999 * 172.21305 * 6 5 4
16 H 1.10009 * 123.03220 * 166.97475 * 6 5 15
( 1, 0) 1.690 2.190 40.715054 0.003 0.11
( 1, 1) 1.690 2.340 35.569649 0.015 0.08
( 0, 1) 1.540 2.340 29.284653 0.018 0.06
( -1, 1) 1.390 2.340 39.472860 0.013 0.08
( -1, 0) 1.390 2.190 46.049811 0.024 0.08
( -1, -1) 1.390 2.040 55.017033 0.021 0.08
( 0, -1) 1.540 2.040 43.383840 0.025 0.05
( 1, -1) 1.690 2.040 47.827091 0.028 0.06
( 2, -1) 1.840 2.040 56.244523 0.005 0.12
( 2, 0) 1.840 2.190 50.784928 0.010 0.09
( 2, 1) 1.840 2.340 46.660710 0.024 0.11
( 2, 2) 1.840 2.490 44.370228 0.019 0.12
( 1, 2) 1.690 2.490 32.564754 0.007 0.06
( 0, 2) 1.540 2.490 25.690229 0.016 0.08
( -1, 2) 1.390 2.490 35.349368 0.012 0.08
( -2, 2) 1.240 2.490 80.022535 0.014 0.11
( -2, 1) 1.240 2.340 84.646107 0.022 0.08
( -2, 0) 1.240 2.190 91.814045 0.026 0.08
( -2, -1) 1.240 2.040 101.426156 0.013 0.09
( -2, -2) 1.240 1.890 111.343212 0.013 0.11
( -1, -2) 1.390 1.890 64.209700 0.014 0.06
( 0, -2) 1.540 1.890 51.534253 0.016 0.08
( 1, -2) 1.690 1.890 54.293939 0.005 0.11
( 2, -2) 1.840 1.890 59.588034 0.012 0.48
( 3, -2) 1.990 1.890 59.881834 0.003 0.14
( 3, -1) 1.990 2.040 62.164030 0.009 0.12
( 3, 0) 1.990 2.190 59.758125 0.004 0.11
( 3, 1) 1.990 2.340 57.177230 0.030 0.11
( 3, 2) 1.990 2.490 55.843144 0.014 0.09
( 3, 3) 1.990 2.640 55.294109 0.022 0.16
( 2, 3) 1.840 2.640 43.183646 0.012 0.08
( 1, 3) 1.690 2.640 30.884478 0.018 0.06
( 0, 3) 1.540 2.640 23.594402 0.015 0.09
( -1, 3) 1.390 2.640 32.871171 0.007 0.09
( -2, 3) 1.240 2.640 77.168837 0.004 0.14
( -3, 3) 1.090 2.640 182.248538 0.022 0.17
( -3, 2) 1.090 2.490 185.472372 0.024 0.08
( -3, 1) 1.090 2.340 190.565645 0.014 0.06
( -3, 0) 1.090 2.190 198.268720 0.006 0.12
( -3, -1) 1.090 2.040 208.431594 0.031 0.09
( -3, -2) 1.090 1.890 218.901813 0.022 0.09
( -3, -3) 1.090 1.740 226.074837 0.006 0.12
( -2, -3) 1.240 1.740 118.038537 0.005 0.08
( -1, -3) 1.390 1.740 70.130494 0.018 0.08
( 0, -3) 1.540 1.740 56.087935 0.008 0.11
( 1, -3) 1.690 1.740 56.309746 0.004 0.11
( 2, -3) 1.840 1.740 57.143406 0.013 0.12
( 3, -3) 1.990 1.740 52.820072 0.008 0.12
( 4, -3) 2.140 1.740 46.049449 0.017 0.17
( 4, -2) 2.140 1.890 55.601664 0.021 0.06
( 4, -1) 2.140 2.040 62.735056 0.022 0.33
( 4, 0) 2.140 2.190 65.665425 0.024 0.12
( 4, 1) 2.140 2.340 65.798877 0.026 0.12
( 4, 2) 2.140 2.490 65.888188 0.011 0.14
( 4, 3) 2.140 2.640 66.219719 0.025 0.19
( 4, 4) 2.140 2.790 66.505363 0.026 0.22
( 3, 4) 1.990 2.790 55.012355 0.011 0.11
( 2, 4) 1.840 2.790 42.488275 0.015 0.06
( 1, 4) 1.690 2.790 29.872923 0.002 0.11
( 0, 4) 1.540 2.790 22.320460 0.017 0.14
( -1, 4) 1.390 2.790 31.354082 0.005 0.16
( -2, 4) 1.240 2.790 75.402298 0.010 0.19
( -3, 4) 1.090 2.790 180.217560 0.025 0.16
( -4, 4) 0.940 2.790 380.493231 0.018 0.19
( -4, 3) 0.940 2.640 382.783941 0.008 0.08
( -4, 2) 0.940 2.490 386.341375 0.027 0.06
( -4, 1) 0.940 2.340 391.835651 0.006 0.09
( -4, 0) 0.940 2.190 399.982591 0.022 0.08
( -4, -1) 0.940 2.040 410.595954 0.017 0.11
( -4, -2) 0.940 1.890 421.512522 0.011 0.12
( -4, -3) 0.940 1.740 429.028052 0.014 0.16
( -4, -4) 0.940 1.590 431.820615 0.012 0.25
( -3, -4) 1.090 1.590 229.264350 0.010 0.12
( -2, -4) 1.240 1.590 121.315767 0.003 0.39
( -1, -4) 1.390 1.590 73.038903 0.010 0.11
( 0, -4) 1.540 1.590 57.847478 0.021 0.12
( 1, -4) 1.690 1.590 55.724873 0.006 0.14
( 2, -4) 1.840 1.590 53.335667 0.030 0.14
( 3, -4) 1.990 1.590 46.578536 0.020 0.19
( 4, -4) 2.140 1.590 38.386999 0.007 0.22
( 5, -4) 2.290 1.590 31.876984 0.023 0.22
( 5, -3) 2.290 1.740 40.473543 0.015 0.09
( 5, -2) 2.290 1.890 51.376117 0.020 0.09
( 5, -1) 2.290 2.040 60.809923 0.019 0.09
( 5, 0) 2.290 2.190 67.902022 0.003 0.12
( 5, 1) 2.290 2.340 72.161126 0.017 0.14
( 5, 2) 2.290 2.490 74.504123 0.019 0.16
( 5, 3) 2.290 2.640 76.102095 0.012 0.20
( 5, 4) 2.290 2.790 77.174429 0.015 0.22
( 5, 5) 2.290 2.940 77.837837 0.014 0.25
( 4, 5) 2.140 2.940 66.666740 0.016 0.09
( 3, 5) 1.990 2.940 54.814947 0.023 0.06
( 2, 5) 1.840 2.940 42.036639 0.022 0.12
( 1, 5) 1.690 2.940 29.236316 0.011 0.16
( 0, 5) 1.540 2.940 21.539397 0.012 0.47
( -1, 5) 1.390 2.940 30.443266 0.012 0.12
( -2, 5) 1.240 2.940 74.352055 0.014 0.17
( -3, 5) 1.090 2.940 179.001739 0.026 0.20
( -4, 5) 0.940 2.940 379.090219 0.011 0.25
( -5, 5) 0.790 2.940 720.524119 0.018 0.23
( -5, 4) 0.790 2.790 722.098428 0.008 0.08
( -5, 3) 0.790 2.640 724.584242 0.023 0.06
( -5, 2) 0.790 2.490 728.360388 0.004 0.11
( -5, 1) 0.790 2.340 734.092018 0.024 0.11
( -5, 0) 0.790 2.190 742.488655 0.012 0.12
( -5, -1) 0.790 2.040 753.379220 0.019 0.11
( -5, -2) 0.790 1.890 764.647132 0.021 0.14
( -5, -3) 0.790 1.740 772.282139 0.009 0.28
( -5, -4) 0.790 1.590 521.937851 0.015 0.42
( -5, -5) 0.790 1.440 523.243627 0.014 0.62
( -4, -5) 0.940 1.440 223.363108 0.020 0.08
( -3, -5) 1.090 1.440 65.779076 0.015 0.08
( -2, -5) 1.240 1.440 1.769176 0.009 0.08
( -1, -5) 1.390 1.440 -5.174536 0.021 0.11
( 0, -5) 1.540 1.440 18.250897 0.015 0.12
( 1, -5) 1.690 1.440 53.416885 0.035 0.14
( 2, -5) 1.840 1.440 89.180271 0.019 0.17
( 3, -5) 1.990 1.440 120.319173 0.022 0.30
( 4, -5) 2.140 1.440 141.573508 0.023 0.27
( 5, -5) 2.290 1.440 155.830031 0.020 0.51
HORIZONTAL STEP= 0.150 ON COORD. 1 OF ATOM 4
VERTICAL STEP= 0.150 ON COORD. 1 OF ATOM 6
UPPER LEFT CORNER COORDINATES: 0.790 2.940 AT (-5, 5)
LOWER RIGHT CORNER COORDINATES: 2.290 1.440 AT ( 5,-5)
THE GRID IS RELATIVE TO THE CENTRAL HEAT OF FORMATION= 35.205941
WHOLE OF (11x11) GRID (Kcal/Mol), SUITABLE FOR PLOTTING:  5 685.32 343.88 143.80 39.15 -4.76 -13.67 -5.97 6.83 19.61 31.46 42.63
4 686.89 345.29 145.01 40.20 -3.85 -12.89 -5.33 7.28 19.81 31.30 41.97
3 689.38 347.58 147.04 41.96 -2.33 -11.61 -4.32 7.98 20.09 31.01 40.90
2 693.15 351.14 150.27 44.82 0.14 -9.52 -2.64 9.16 20.64 30.68 39.30
1 698.89 356.63 155.36 49.44 4.27 -5.92 0.36 11.45 21.97 30.59 36.96
0 707.28 364.78 163.06 56.61 10.84 0.00 5.51 15.58 24.55 30.46 32.70
-1 718.17 375.39 173.23 66.22 19.81 8.18 12.62 21.04 26.96 27.53 25.60
-2 729.44 386.31 183.70 76.14 29.00 16.33 19.09 24.38 24.68 20.40 16.17
-3 737.08 393.82 190.87 82.83 34.92 20.88 21.10 21.94 17.61 10.84 5.27
-4 486.73 396.61 194.06 86.11 37.83 22.64 20.52 18.13 11.37 3.18 -3.33
-5 488.04 188.16 30.57 -33.44 -40.38 -16.96 18.21 53.97 85.11 106.37 120.62
-5 -4 -3 -2 -1 0 1 2 3 4 5
STATIONARY POINT(s) LOCATED ON THE GRID:
5 685.32 343.88 143.80 39.15 -4.76 -13.67 -5.97 6.83 19.61 31.46 42.63
4 686.89 345.29 145.01 40.20 -3.85 -12.89 -5.33 7.28 19.81 31.30 41.97
3 689.38 347.58 147.04 41.96 -2.33 -11.61 -4.32 7.98 20.09 31.01 40.90
2 693.15 351.14 150.27 44.82 0.14 -9.52 -2.64 9.16 20.64 30.68 39.30
1 698.89 356.63 155.36 49.44 4.27 -5.92 0.36 11.45 21.97 30.59 36.96
0 707.28 364.78 163.06 56.61 10.84 0.00 5.51 15.58 24.55 30.46 32.70
-1 718.17 375.39 173.23 66.22 19.81 8.18 12.62 21.04 26.96 27.53 25.60
-2 729.44 386.31 183.70 76.14 29.00 16.33 19.09 24.38 24.68 20.40 16.17
-3 737.08 393.82 190.87 82.83 34.92 20.88 21.10 21.94 17.61 10.84 5.27
-4 486.73 396.61 194.06 86.11 37.83 22.64 20.52 18.13 11.37 3.18 -3.33
-5 488.04 188.16 30.57 -33.44 -40.38 -16.96 18.21 53.97 85.11 106.37 120.62
-5 -4 -3 -2 -1 0 1 2 3 4 5
STATIONARY POINT(s) LOCATED ON THE GRID:  SADDLE-POINT ( 1,-4) HEAT OF FORMATION= 55.7249
ATOM CHEMICAL BOND LENGTH BOND ANGLE TWIST ANGLE
NUMBER SYMBOL (ANGSTROMS) (DEGREES) (DEGREES)
(I) NA:I NB:NA:I NC:NB:NA:I NA NB NC
1 C
2 C 1.44145 * 1
3 C 1.41309 * 119.41629 * 2 1
4 C 1.69000 104.90958 * -60.57540 * 3 2 1
5 C 1.41309 * 104.90971 * 53.77858 * 4 3 2
6 C 1.59000 107.40738 * 59.88959 * 1 2 3
7 H 1.11757 * 113.19137 * 179.15859 * 1 2 3
8 H 1.12010 * 113.36283 * 124.49776 * 1 2 7
9 H 1.09715 * 119.78836 * -256.90159 * 2 3 4
10 H 1.11265 * 106.18820 * 176.59417 * 3 4 5
11 H 1.11504 * 101.73155 * -67.71076 * 3 4 5
12 H 1.11265 * 106.18903 * 176.59402 * 4 3 2
13 H 1.11504 * 101.73102 * -67.71164 * 4 3 2
14 H 1.09715 * 119.78726 * 103.10206 * 5 4 3
15 H 1.11757 * 113.19016 * 179.15665 * 6 5 4
16 H 1.12010 * 113.36416 * 124.49723 * 6 5 15
MAXIMUM (-5,-3) HEAT OF FORMATION= 772.2821
ATOM CHEMICAL BOND LENGTH BOND ANGLE TWIST ANGLE
NUMBER SYMBOL (ANGSTROMS) (DEGREES) (DEGREES)
(I) NA:I NB:NA:I NC:NB:NA:I NA NB NC
1 C
2 C 1.38689 * 1
3 C 1.62957 * 115.73594 * 2 1
4 C 0.79000 124.93220 * -33.17518 * 3 2 1
5 C 1.62957 * 124.93384 * 17.66387 * 4 3 2
6 C 1.74000 102.65454 * 47.99306 * 1 2 3
7 H 1.11176 * 116.44124 * 166.28804 * 1 2 3
8 H 1.11378 * 118.91585 * 136.05828 * 1 2 7
9 H 1.08656 * 116.35604 * -229.35206 * 2 3 4
10 H 1.22611 * 124.34624 * 144.69452 * 3 4 5
11 H 1.23931 * 125.32798 * -98.35098 * 3 4 5
12 H 1.22612 * 124.34562 * 144.69504 * 4 3 2
13 H 1.23931 * 125.32748 * -98.35228 * 4 3 2
14 H 1.08656 * 116.35546 * 130.65323 * 5 4 3
15 H 1.11176 * 116.44162 * 166.28509 * 6 5 4
16 H 1.11379 * 118.91430 * 136.05813 * 6 5 15
ELAPSED WALL CLOCK TIME : 6.42 SECONDS
FULL COMPUTATION TIME : 23.32 SECONDS
SADDLE-POINT ( 1,-4) HEAT OF FORMATION= 55.7249
ATOM CHEMICAL BOND LENGTH BOND ANGLE TWIST ANGLE
NUMBER SYMBOL (ANGSTROMS) (DEGREES) (DEGREES)
(I) NA:I NB:NA:I NC:NB:NA:I NA NB NC
1 C
2 C 1.44145 * 1
3 C 1.41309 * 119.41629 * 2 1
4 C 1.69000 104.90958 * -60.57540 * 3 2 1
5 C 1.41309 * 104.90971 * 53.77858 * 4 3 2
6 C 1.59000 107.40738 * 59.88959 * 1 2 3
7 H 1.11757 * 113.19137 * 179.15859 * 1 2 3
8 H 1.12010 * 113.36283 * 124.49776 * 1 2 7
9 H 1.09715 * 119.78836 * -256.90159 * 2 3 4
10 H 1.11265 * 106.18820 * 176.59417 * 3 4 5
11 H 1.11504 * 101.73155 * -67.71076 * 3 4 5
12 H 1.11265 * 106.18903 * 176.59402 * 4 3 2
13 H 1.11504 * 101.73102 * -67.71164 * 4 3 2
14 H 1.09715 * 119.78726 * 103.10206 * 5 4 3
15 H 1.11757 * 113.19016 * 179.15665 * 6 5 4
16 H 1.12010 * 113.36416 * 124.49723 * 6 5 15
MAXIMUM (-5,-3) HEAT OF FORMATION= 772.2821
ATOM CHEMICAL BOND LENGTH BOND ANGLE TWIST ANGLE
NUMBER SYMBOL (ANGSTROMS) (DEGREES) (DEGREES)
(I) NA:I NB:NA:I NC:NB:NA:I NA NB NC
1 C
2 C 1.38689 * 1
3 C 1.62957 * 115.73594 * 2 1
4 C 0.79000 124.93220 * -33.17518 * 3 2 1
5 C 1.62957 * 124.93384 * 17.66387 * 4 3 2
6 C 1.74000 102.65454 * 47.99306 * 1 2 3
7 H 1.11176 * 116.44124 * 166.28804 * 1 2 3
8 H 1.11378 * 118.91585 * 136.05828 * 1 2 7
9 H 1.08656 * 116.35604 * -229.35206 * 2 3 4
10 H 1.22611 * 124.34624 * 144.69452 * 3 4 5
11 H 1.23931 * 125.32798 * -98.35098 * 3 4 5
12 H 1.22612 * 124.34562 * 144.69504 * 4 3 2
13 H 1.23931 * 125.32748 * -98.35228 * 4 3 2
14 H 1.08656 * 116.35546 * 130.65323 * 5 4 3
15 H 1.11176 * 116.44162 * 166.28509 * 6 5 4
16 H 1.11379 * 118.91430 * 136.05813 * 6 5 15
ELAPSED WALL CLOCK TIME : 6.42 SECONDS
FULL COMPUTATION TIME : 23.32 SECONDS
|
This is a description of the center point of the grid, including heat of formation and gnorm. |
|
|
This is the geometry of the center point of the grid. |
|
|
This is a list of the AM1 energies of the optimized geometries at each pair of STEP1, STEP2 coordinates. Due to the size of the output, the geometries
are not printed. Coordinates and information about each geometry can be found in the
visualization ( |
|
|
This is an 11×11 matrix of the energies at the points in the order described above. AGUI can present this data in graphical form. |
|
|
A list of (constrained) critical points and geometries that were found during the grid search. Constrained critical points may not actually correspond to a true critical point. Critical points of interest should be reoptimized with all optimization flags set to on and a FORCE calculation performed on the resulting geometry. |
|
Copyright © 1992-2013 Semichem, Inc. All rights reserved. |
|
|
 |
|
| Chapter 25. One-Dimensional Reaction Pathway |  |
Chapter 27. Finding a Specific Transition State (CHN) |