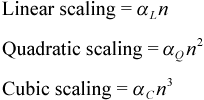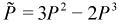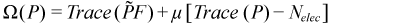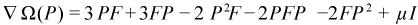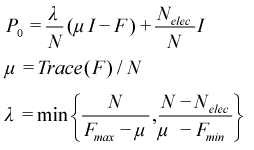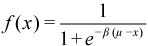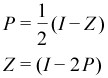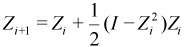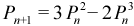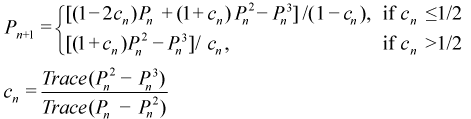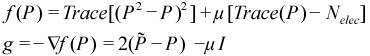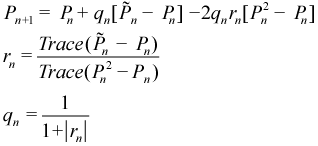| Chapter 7. Sparse Matrix Methods | ||
|---|---|---|

|
Part I. AMPAC™ 10 User Guide |  |
| Chapter 7. Sparse Matrix Methods | ||
|---|---|---|
|
|
Part I. AMPAC™ 10 User Guide | |
Table of Contents
As the field of quantum chemistry continues to grow and evolve, there is an increasing desire to study larger and larger chemical systems. In particular, complex biochemical systems with thousands and tens of thousands of atoms are quickly becoming the new frontier for quantum chemical investigation. Semi-empirical methods are very fast yet accurate, so are an excellent candidate for this type of study. One major hurdle, however, is that the amount of computational effort required to perform the calculation grows very rapidly as you move to larger and larger systems until it becomes prohibitively expensive. Traditionally, the Fock matrix build is the slowest and most expensive step in a calculation but even if we were able to speed that up, there will still come a point where even the most basic matrix operations, such as diagonalization or matrix-matrix multiplication, will consume so much time that it prevent us from going to larger systems. The amount of CPU time is not the only factor that limits the size of traditional semiempirical calculations. In many cases, the amount of memory required to run a very large molecule is an even more severe constraint on size. Only by lifting these two very fundamental limitations can we handle calculations larger than we could have dreamed of before. This section introduces new AMPAC functionality designed to meet this goal.
The heart of the issue when it comes to studying larger molecues is how fast do the computational requirements (primarily CPU time and memory needed) grow as a function of the size of the system. This is known as the computational scaling for that particular method. If we understand this scaling, then we can project approximately how long a job will take and how much memory will be required and hence the determine the largest job feasible for a given computer. The actual computational scaling is an extremely complex function that depends on the details of the jobs, the particular computer, the operating system, etc. so is not very helpful. Once the molecule gets large enough, however, the cost is dominated by just a few key steps allowing us to reduce the scaling to a fairly simple function, which makes the resulting analsis much more manageable. This asymptotic behavior can then be used to evaluate the performance of the method and to compare it against other methods.
The holy grail for studying large chemical systems is "linear scaling methods," where the computational requirements
(in the asymptotic limit) only increase linearly with system size. To understand what that means, we need to define some
terms. When we talk about the size of the system in this context, we typically use the number of atoms or better yet the
number
of basis functions as the independent variable, n. The amount of CPU or memory required is
then expressed as a function of this variable. For our purposes here, we need only consider simple polynomial powers of
n, namely cubic, quadratic, and linear scaling.
For this simple example, we can consider how rapidly in cost each as the function of system size until they hit some
user-defined limit representing the maximum amount of resource (i.e. CPU time or memory) allowed for a calculation.
The value of n at which we reach this limit then represents the largest system of that type
allowable for a program with that scaling. In general, of course, we expect a method with cubic scaling will hit this limit
first for relatively small n, while linear scaling allows the largest systems.
There is an important caveat to this analysis. In looking at the scaling function, we must also consider
the coefficient in front of the polynomial. While linear scaling will ultimately always be better
than cubic or quadratic scaling, it will generally not be optimal for all values of n.
This gives rise to the concept of "crossover"--the point at which linear scaling becomes better (i.e. has a lower cost).
The importance to the user is that the sparse matrix methodology described below will only be helpful for systems that
are large enough to truly benefit from using this approach. At the same time, we should remember that this
scaling concept applies independently to the CPU time required and the memory limit, so there may be cases
where our linear scaling method is superior to the traditional method for one of these requirements but not both.
The traditional way that matrices are stored is referred to as "full matrix" because it includes all matrix elements even if they are zero. For extremely large molecues, the atoms are spread out in space so that interactions between pairs of widely separated atoms quickly become negligible. In these cases, the total number of important elements may only grow linearly with the size of the system but the full matrix approach cannot take advantage of this because it must store all elements, which grows quadratically with system size. The way to exploit this feature is known as "sparse matrices," where you only the store the non-negligible components of the matrix. In addition to the actual numerical data, sparse matrices require two additional data structures to keep track of where each element belongs in the matrix. With all of this information, one can represent the original full matrix but with storage that is proportional only to the number of non-negligible elements. (We use a very generic sparse matrix format for our work although more sophisticated forms exist for certain specialized problems.)
Reducing matrix storage is one thing but we still need to do all of the necessary operations using these sparse matrices. Temporarily converting the sparse matrices to full matrices and then performing the operation using standard matrix routines would defeat the purpose (because it would reintroduce the full matrix bottleneck) and so is not a viable option. Much work has been done to develop sparse matrix equivalents to standard matrix-vector and matrix-matrix type operations that take full advantage of the sparse matrix structure. While these routines are generally not as efficient as traditional full matrix routines, they do benefit from only acting on the stored elements. The CPU time for a simple matrix-matrix multiplication using full matrices will scale cubically with the size of the system. Using sparse matrices, however, the CPU cost may only go up linearly for large enough systems. So, sparse matrices have the potential to fix both the storage space and CPU cost bottleneck problems caused by full matrices.
Even with that, several major challenges still remain. The most critical one is finding a way to solve for the molecule's density matrix, which is needed to determine the molecule's energy, gradient, and other essential properties. Normally, this is done by iteratively diagonalizing the Fock matrix to get a set of molecular orbitals, which are then used to form the density matrix; but, unfortunately there is no sparse matrix equivalent to diagonalization. A number of iterative procedures have been developed to solve for the density using only sparse matrix operations, which will be described in the next section. This leads to the second challenge, which is insuring that the resulting density matrix satisfies N-representabllity (i.e. represents the correct number of electrons and is idempotent). When the density matrix deviates from these properties, this must be corrected by a process known as purification. The third major issue involves determining which matrix elements to keep and which to negelect in reliable but efficient manner. Strategies for dealing with these two later problems are discussed in their own separate sections below.
Solving for the self-consistent density matrix lies at the very heart of semiempirical methods, so it is critical that this is done in a way that is fast, accurate, and reliable. AMPAC's normal SCF engine cannot be used with sparse matrices, so new iterative methods were implemented to handle this case. Currently, there are five different solvers to choose from, each with its own strengths and weaknesses. (While these are designed specifically for sparse matrix jobs, they can also be run in full matrix mode. Running these using full matrices is expensive for very large molecules just like standard SCF but may be helpful for testing purposes. In other words, this can be used to test the algorithmic issues separately from sparse matrix ones.)
PSOLVE=CGDMS. The basic strategy of the conjugate-gradient density matrix search (CG-DMS)[20] [21] [22] is to express the energy as a functional of electron density and then minimize it. We start by considering the basic energy functional: Trace(PF), where P is the density and F is the Fock matrix. Next, we must consider that the density matrix has properties that must be maintained throughout the minimization, namely the correct number of electrons and idempotency. (These critical properties are discussed in more detail in a subsequent section.) In order to avoid doing a constrained optimization, we need to modify this simple functional in a way that will naturally preserve these properties. The first modification is that we replace the density in the energy portion of the functional with the McWeeny purified density matrix expression:
This helps preserve idempotency during the optimization. The second change is to add a Lagrangian term to preserve the number of electrons. Doing this, we arrive at the CGDMS functional and its gradient:
The value of μ is chosen such that the gradient is traceless, which insures that line searches preserve the trace of the density. The actual search step is pre-conditioned (unless NOPRECON) using the inverse of the diagonal Hessian (hence this is a quasi-Newton step). In certain rare cases, elements of the diagonal Hessian may become small or negative, which must be avoided using a level shift technique. Level shifting usually happens automatically when needed but the value for the shift can be manually specified using VSHIFT. From here, we do an unconstrained energy minimization using the conjugate-gradient (CG) method. (We use the Polak-Ribere form of CG by default but the Fletcher-Reeves version can be specified by keyword FLETCHER.) The step size for the CG line search is determined analytically by solving a cubic equation. Normally, CG iterations should always be fully converged but for our case this is wasteful and we typically need to take no more than 4 CG steps per SCF cycle. This is strategy is known as progressive convergence but can be turned off in favor of fully converging the density every SCF cycle using keyword FULLCNV. For smooth convergence, DIIS (Direct Inversion of the Iterative Subspace) is applied during the CG procedure. This is known as CG-DIIS and is distinct from SCF-DIIS, which is applied at the SCF level. (The CGDIIS keyword controls the behavior of CG-DIIS.)
PSOLVE=QNDMS. QN-DMS stands for quasi-Newton density matrix search.[23] This is essentially the same as CG-DMS except that the analytic line-search step is replaced by a simple inexpensive quasi-Newton step to make things faster. To compensate, CG-DIIS is used to find the best step as a linear combination of previous steps. (CG-DIIS is optional with CG-DMS but required with QN-DMS.) When successful, QN-DMS can be as much as 30% faster than CG-DMS although it may not be quite as reliable.
PSOLVE=PDM. Purification of the density matrix (PDM)[24] is the simplest and often the most efficient of all the currently available methods. The first step is to produce an initial density from the Fock matrix of the form:
The value λ is chosen such that it has eigenvalues of the resulting density matrix are in the range one to zero, while the form of the equation guarantees that it has the correct trace (i.e. correct number of electrons). This density is not idempotent, so up to 20 cycles of purification are used to make it idempotent. Determining λ requires that we first determine the maximum, Fmax, and minimum, Fmin, eigenvalues of the Fock matrix. By default, AMPAC uses a simple eigensolver to compute these values. If this procedure fails or if the keyword GERSH is present, then the Gershgorin theorem is used to generate bounds on the eigenvalues instead. The Gershgorin bounds are typically much wider than the real limits, so the resulting initial density requires more purification cycles to achieve idempotency.
PSOLVE=CEM. A more sophisticated variant of the PDM method is the Chebyshev expansion method or CEM.[25] [26] The core idea is that the density matrix can obtained from the Fock matrix and a Fermi-Dirac distribution for the orbital occupancies. The Fermi-Dirac distribution is constructed to map occupied eigenvalues to one and virtual ones to zero and is expressed as:
The value of ϐ is fixed at 100 and μ is determined such that the resulting density will have the correct number of
electrons. This Fermi-Dirac function is expanded in terms of Chebyshev polynomials and truncated to a finite order.
Chebyshev polynomials are defined over the range [-1:1], so the Fock matrix must be scaled and shifted so that its eigenvalues
fall within this range. To do that, we need to determine the span of the Fock matrix eigenvalues, which is accomplished
by actually solving for minimum and maximum eigenvalues or using the Gershgorin bounds (if keyword
GERSH) along the same lines of PDM. The density matrix
is then constructed as a Chebyshev power series using the scaled/shifted Fock matrix. (Since the density matrix is just
a linear combination of powers of the Fock matrix that means that it will share the same eigenvectors as the Fock matrix.)
If the expansion contains enough terms, this will precisely reproduce the correct idempotent density. Unfortunately, this
is
not very efficient because each additional term in the expansion requires one additional matrix multiplication and addition.
If we reduce the order of the expansion, then the initial density will not be idempotent so that we have to apply density
matrix purification
(similar to what is done in PDM) to compensate. So, there is a trade-off that needs to be considered here. A large Chebyshev
expansion requires the most computational effort and memory to produce the initial density but is the most stable and reliable.
A minimal (second order) expansion has the lowest cost (with most of the effort put into purifying the initial density)
and is
very similar in spirit and performance to PDM. The default behavior is to truncate the Chebyshev expansion at order 5,
which
is a good compromise between reliability and cost. If CEM encounters trouble, try increasing the order of the expansion
using
PSOLVE=CEM=n.
PSOLVE=SIGN. The Sign Matrix method (SIGN)[27] solves for the sign matrix, which is then converted into the density matrix. A sign matrix, Z, has eigenvalues -1 and +1 as opposed to the density matrix, P, which has eigenvalues of +1 and 0. Given this, we can easily convert between these two types of matrices.
The principle benefit of the sign matrix is that one can use Newton-Schultz iterations (NSI) to drive an approximate sign matrix to an exact one in the same manner that purification does for the density matrix.
This standard NSI has been modified and adapted to fit our specific needs as will be explained in more detail in a subsequent section. The SIGN method begins by determining the upper and lower bounds of the Fock matrix and then scaling it to produce an approximate initial sign matrix (analogous to PDM). NSI is applied to converge it to a pure sign matrix and this is then converted into the corresponding density matrix. The notable parallels between SIGN and PDM are in fact nearly exact. If we convert each sign matrix expression to use the more convenient density matrices using the appropriate identity, the result is identical to PDM except that the modified NSI is used for the purification step instead of the generalized purification. This is discussed in more detail in the next section.
All Methods. The names for each of these methods refer to the overall strategy but also refers to the method used during an individual SCF step. This subtle distinction is important because in certain cases more than one method may be used in the course of solving for the density. PSOLVE=CGDMS or QNDMS, for example, may use several iterations of PDM for the initial steps of the SCF. Also if either of these two methods encounter certain unresolvable difficulties, they may automatically switch to using PDM iterations for remainder of the calculation.
In solving for the density matrix, we must be mindful that it has to remain valid throughout the optimization to insure that we have a physically meaningful solution. In the standard SCF procedures, the density matrix is constructed from a set of orthonormal molecular orbitals, which guarantees that it will properly satisfy N-representablity. In the case of our sparse matrix density solvers, this is much more complicated for two reasons. First, algorithms themselves necessarily perturb the density and must then restore it to a valid state. Second, the fact that we must truncate small elements along the way is yet another approximation. Fortunately, there are efficient ways of dealing with this problem.
The density matrix must satisfy two separate conditions that must be simultaneously satisfied. First, the density must have the correct number of electrons, i.e. Trace(P) = Nelec. Enforcing this is easy, because we can simply scale the entire matrix by the necessary factor. The second property is that the density must be idempotent, P2=P. Enforcing this is far more challenging. The basic approach used here is that when P is not idempotent, a new density is generated from it that will be closer to idempotency. This procedure is then repeated iteratively until the idempotency has been restored to an acceptable degree. We refer to this process as "purification." Several different methods for purification are available.
McWeeny purification. McWeeny's formula is elegant, useful, and represents the original approach to sparse matrix density purification.[28]
This formula works by shifting eigenvalues in the range [-0.5,0.5] toward 0, while those in the range [0.5,1.5] will go to 1. When the occupied orbitals reach 1 and virtual ones reach 0, then the matrix satisfies idempotency and the procedure is complete. In the event that P is already idempotent, then the McWeeny purified density simply returns P. It should be noted that if the eigenvalues of the density matrix fall outside the range [-0.5,1.5], each iteration of McWeeny purification will casue the density to become even less idempotent and will eventually cause the calculation to fail.
Generalized purification. A generalization of McWeeny's formula was developed to yield better results but at the same cost.[24]
In the original McWeeny formula, 1/2 represents a fixed point boundary between those eigenvalues that will be converged toward 1 and those that will go to 0. This may lead to poor convergence if the eigenvalues of the approximate density are poorly distributed, which occurs when an eigenvalue is on the opposite side of this fixed point than it should be and so gets driven toward the wrong value. This is a common problem very early in the SCF procedure when the density is far from its final form. Generalized purification treats this dividing line as a variable, c, which is allowed to vary dynamically to better fit the nature of the density. Once the density is very close to convergence, this formula is essentially the same as standard McWeeny and c = 1/2. Generalized purification is usually more reliable and converges in fewer iterations than McWeeny at no extra cost.
Steepest descent purification. For this method, the idempotency and trace of the density matrix is expressed as a simple functional.[29]
The search direction, g, is the negative of the gradient of the functional, which represents a steepest descent direction. The optimal step size is found by analytically solving a simply cubic equation. The primary advantage of this steepest descent approach is that each step is guaranteed to make the density more idempotent. (The McWeeny and generalized purification may diverge in some cases.) The main disadvantage is that the CPU cost is about twice that of the McWeeny purification but with only modest improvement in convergence. The main problem is that it lacks the flexibility of being able to shift the fixed point the way that generalized purification can.
Newton-Shultz iterations. Newton-Schultz iterations (NSI) (see Equation (7.8)) were developed to converge sign matrices (analogous to how McWeeny purifies a density matrix). This standard NSI was modified to preserve the trace of the density (and hence maintain the total number of electrons).[27] For AMPAC, all of the sign matrix work has been directly recast in terms of density matrices. This makes for more efficient computation and can be more readily compared to other purification methods.
In this incarnation, it is similar in spirit to generalized purification in that it allows the fixed point to vary dynamically and performs nearly as well. NSI is only used with PSOLVE=SIGN and is done in place of generalized purification.
The primary difference between full and sparse matrices is that sparse matrices require a symbolic form in addition to its numeric content. The symbolic form of sparse matrix consists of two integer arrays that specify the matrix indices for each of the numeric elements. It is this symbolic form that allows us to store and manipulate only the non-negligable elements of the matrix. This comes at a cost, however, because this form has to generated in addition to the normal work of computing the numerical elements. For simple matrix operations there are procedures by which we can determine the form of the result matrix. Only numeric elements specified in this symbolic form are actually computed, so we don't waste time computing zeros (which is the main point of using sparse matrices).
Most sparse matrix operations occur in two distinct steps: (1) create the symbolic matrix form that describes the locations all of the potentially non-zero elements and then (2) fill in the specified numeric elements. (Some negligible elements may formed by due to numeric cancellation, but these can be trivially weeded out once the matrix has been formed.) This insures that the matrix has enough room for all of the necessary elements. We refer to this strategy as "Let It Grow," because the form is constructed to meet the needs of the operation. This works well enough for most problems but is inefficient for the density matrix solve and certain other calculations. There are two issues at work here. First, the computational cost of determining the matrix form is almost as much as it is for the actual filling in the numeric values. Second, the matrix form will grow from cycle to cycle (even after we eliminate negligible elements), causing a loss of linear scaling in both memory and CPU. (The sparse matrix work here is different from typical sparse matrix usage in that the matrix form is defined by a neglect threshold rather than being exactly defined by the nature of the problem. As a result, "Let It Grow" may result in far too many unnecessary elements and can be very inefficient.)
A "Fixed Form" strategy has been developed for use within the density solving routines. The basic idea is that during an individual SCF cycle, the density, Fock, and other intermediate matrices are given a common form. Since the symbolic form is then known, only the numeric operations need to be performed resulting in a significant savings in CPU. A single matrix form is used, rather than one per matrix, resulting in a memory savings as well. Most importantly, the matrix form is prevented form expanding. The matrix form is updated each SCF cycle, so it is allowed to grow between cycles but not within the cycle. The central challenge to this approach is to correctly determine the common symbolic form to use. If it is chosen too few elements, the density solver may perform poorly or fail. If it is chosen too many, the procedure will be inefficient. The current strategy is to use combined form of P, F, PF, and FP, where P is the density and F is the Fock matrix for that cycle. In certain cases, this may not be sufficient and keyword ENHANCE can be used to include more elements in the fixed form, which may alleviate the problem. When this keyword is used, the form of PFP is included for CGDMS or QNDMS or P02 for PDM, CEM, or SIGN (where P0 is corresponds to Equation (7.5)).
Running a sparse matrix calculation involves a number of important trade-offs and other issues that need to be carefully considered before running a job. The first and most important is whether or not to use sparse matrices at all. Unfortunately, sparse matrices are not a cure-all because the degree to which a particular molecule can benefit from this approach depends considerably on the geometry of the system. A very compact and tightly bound structure will limit the number of interactions that can be safely ignored and so nullify most of the advantage of sparse matrices. So, even cases with the same number and types of atoms may differ considerably in performance depending on how tightly or loosely they are held together. (Full matrix calculations, in contrast, generally perform the same regardless of the compactness of the structure.) Moreover, sparse matrix operations are generally much less computationally efficient than their full matrix cousins, so only if enough elements can be successfully ignored (which depends on the size and shape of the molecule) will there be enough benefit from using sparse matrices to make it competitive with normal full matrix approaches. As such, using sparse matrices with small and medium sized molecules is generally not recommended. Some experimenting may be necessary to determine if your system of interest will work well enough.
The next major consideration involves the required level of accuracy, i.e. how closely do you need to match the equivalent full matrix calculation for that molecule. The sparse matrix approach involves a critical trade off between accuracy and performace, where you sacrifice one to gain more of the other. This is controlled via the sparse matrix neglect threshold, which is set by keyword SPARSE. Elements in every matrix (density, Fock, overlap, etc.) will be neglected (treated as zero) if it falls below this value. The larger this negelect threshold is, the fewer things need to be stored and less work needs to be done, which makes the calculation faster but less accurate. Chemical studies that do not require as high of a degree of accuracy are more amenable to the sparse matrix approach because they naturally allow a larger neglect threshold.
A neglect threshold of 1 × 10-8 (SPARSE=TIGHT) is generally sufficient to reproduce the full matrix result to high precision. Using threshold smaller than this is generally unnecessarily because it will only make the job run longer. Using 1 × 10-5 (SPARSE=LOOSE) is close to the limit of how large the negelect threshold can be and still achieve convergence. This value will generally give results that differ from full matrix by milli-Hartree, which is excellent for quick exploratory studies. Values between these two limits can be considered based on the project needs and the available resources.
A third factor is the type of job(s) that is being considered. Currently, only the energy, gradient and a few other properties can take full advantage of the sparse matrix paradigm. Geometry optimization, frequencies, solvation, and similar job types will run successfully with sparse matrices turned on but may run very slowly or take up too much memory because these still use full matrices at many critical points. Future works is aimed at removing these limitations but for now the user must be aware of this restriction. A few features, like C.I. (including implicit cases like RHF DOUBLET or EXCITED) will not work at all with sparse matrices. A listing of dependency information is given below.
The last major issue is the choice of solver. There are five density matrix solvers to choose from using the PSOLVE keyword. The results from each are generally quite comparable but they do differ in terms of efficiency and stability. So, if one method fails or does poorly, a different method is likely to succeed.
Given that sparse matrices provides the capability to determine the energy and properties of very large molecues, what about geometry optimization? Being able to optimize structures is obviously a critical feature. For example, if one adds hydrogens to a large PDB file, it is important to be able to relax them before doing an energetics study. To this end, much work has been done in AMPAC to be able to extend the reach of our existing optimizers. There are two main bottlenecks that must be considered: CPU and memory. I will consider each of these in turn.
The main challenge in terms of CPU is eliminating diagonalization, which scales cubically with the number of atoms. For small systems (under 500 atoms) this is fairly fast, but for larger molecules it is simply prohibitive. For TRUSTE there are two main places that diagonalization is traditionally used during the optimization. The first is in the formation of the trust radius step. In AMPAC, this diagonalization is circumvented by using a quadratically scaling iterative procedure--used automatically for systems with more than about 50 atoms. The second use of diagonalization occurs in the formation of the initial guess for the Hessian. Once the guess is generated, it is diagonalized to form the Hessian eigenvalues and eigenvectors. This information is then used to rebuild the Hessian after carefully adjusting any small Hessian eigenvalues that would interfere with the optimization. Most optimizers require the Hessian eigenvalues and eigenvectors as part of their operation and so diagonalization cannot be avoided for these cases. The exception to that rule is TRUSTE but by default the Hessian is still diagonalized to insure a quality Hessian (with no tiny eigenvalues). This behavior can be avoided by using the new variant, TRUSTE=LARGE. This keyword uses the Lindh initial guess Hessian (LINDH) which is naturally positive definite but also uses a different (non-diagonalization) strategy to eliminate small Hessian eigenvalues. The resulting quality of the Hessian is slightly worse (i.e. more geometry steps required) but is necessary for handling truly large molecules.
Storage of the Hessian matrix, its associated eigenvectors, and related matrices are the primary memory bottleneck for large scale optimizations, since this scales quadratically with the number of atoms. Unfortunately, the Hessian is highly non-sparse even for very large spread-out systems, so the sparse matrix strategies employed in calculating the energy cannot be used here. Instead, our strategy has been to eliminate any temporarily allocated quadratic storage, such as occurs with projecting translation/rotation out of the Hessian or converting it from Cartesian to internal coordinates. By eliminating this extra memory consumption to the bare minimum, TRUSTE=LARGE, is now applicable to much larger molecules than previously possible.
TRUSTE=LARGE opens a new door into AMPAC's ability to handle very large molecules. For molecules up to about 5,000 atoms that are that are sufficiently sparse to be handled by a given computer system, it should generally still be optimizable using this keyword. For very sparse systems on computers with a lot of memory, molecules as big as 10,000 atoms are potentially feasible. Beyond that limit is currently unreachable by AMPAC but work is underway to develop a new optimizer capable of handling even those large molecules.
For the most part, running a job with sparse matrices is the same as running a normal full matrix calculation. Just put in your SPARSE keyword and possibly PSOLVE option and you are ready to go. The results should correspond closely to the equivalent full matrix job and only the time and resources required should be significantly different. The majority of the difference between full and sparse happens behind the scenes and is all automated. There are, however, some key differences that the user should be aware of, which I will outline here.
Integral Storage Traditionally, AMPAC will compute and store all of the necessary integrals at the beginning of the calculation and reuse them each SCF iteration as needed. This saves CPU time but increases the amount of memory required. While this is very beneficial for smaller molecules, the memory requirement becomes prohibitive for really large ones. For sparse calculations (or by using the keyword DIRECTSCF), these integrals will not be stored and will be recomputed as needed.
Initial Guess Density. Before we can begin solving for the SCF energy, we need to have an initial guess density to start the procedure. The standard guess is simply a diagonal density matrix, which is generally more than sufficient for most calculations. This actually works well for most sparse matrix cases but sometimes a better guess would be helpful and sometimes is critical. There is an alternate initial guess procedure activated by the keyword LEWIS that tries to determine how the electrons distribute themselves into orbitals and bonds--roughly equivalent to the concept of the Lewis dot structure. From this, it is able to generate an inital density that takes into account the detailed structure of the molecule.
Density Solvers. This is the most time consuming and difficult part of the calculation. There are five different methods (described earlier) to choose from and can be specified using the PSOLVE keyword. The default choice is PDM.
HOMO/LUMO Orbitals. One of the most fundamental ways that sparse matrix calculations differ from traditional ones is that they solve directly for the density matrix, so there are no canonical molecular orbitals. The density is sufficient to determine the energy, gradient, and most other properties but there are a few places where having orbitals and orbital energies are necessary. Finding all of the canonical molecular orbitals is not feasible in a sparse matrix calculation but we can efficiently solve for a small subset orbitals around the HOMO-LUMO gap. This is done using a special routine based on Davidson diagonalization. From this, we can calculate the ionization potential and the HOMO-LUMO energy gap as well as to display the coefficients for these orbitals. This also enables the calculation of hyperfine coupling constants (HYPER). This determination of the HOMO and LUMO orbitals can be skipped to save time by using the NOHL keyword but these specific results would consequently be unavailable.
Lowdin Transformation. Given the overlap matrix, S, the Lowdin transformation is simply S-1/2. For full matrices, this is calculated in the usual way via diagonalizing the overlap matrix. Diagonalization cannot be used with sparse matrices, so an alternative approach had to be developed. The basic strategy is to express the Lowdin transform as a Chebyshev expansion of 1/sqrt(S). While this works, it can be quite slow for large systems.
Matrix Printing. When AMPAC prints a full matrix, all of the elements are shown in a simple dense format. An alternative style of printing matrices had to be developed to include only the non-zero elements of a sparse matrix. Correspondingly, each element has to be individually labeled since we cannot simply examine its position in the matrix. For smaller systems, this style will take up more space in the output file but for larger cases it will save quite a bit of room.
Matrix Storage. Internally, sparse matrices are stored in a
special format to facilitate efficient calculation but this is all hidden from the users. When a sparse
matrix is stored in the .vis file, it must be stored in its
compact sparse matrix form. Whereas a full matrix is stored as a single
continuous array, sparse matrix must stored in four separate arrays labeled IA, JA, AN, AD. IA and JA
are integer arrays that keeps track of the indices of each element. AN and AD store the actual numeric
information, with AD holding the diagonal elements of the matrix and AN holding everything else.
For large sparse systems, the energy and gradient will scale linearly in CPU and memory cost but what about other job types and properties? The good news is that most properties are able to fully exploit sparse matrices and so can be used for the same large systems as the energy. These include BONDS, ENPART, GRADIENTS, HYPERFINE, IADM, LEWIS, MPG, and PI. Only a handful of keywords cannot be used with sparse matrices and these are listed below. The remaining keywords are ones that will run with sparse matrices but do not take full advantage of sparsity and so may not scale linearly with system size. These are also listed below with some comments about their applicability to large systems.
C.I. (direct and indirect) is not allowed. These jobs will not run.
ROHF is not allowed. Currenly, SPARSE is not equipped to run this type of job.
GRAPH will be skipped. The GPT file is not written because there are no molecular orbitals.
QCSCF keyword is ignored. Currently, there is no sparse matrix equivalent of this keyword.
Orbital localization (LOCALIZE, SCFLOCAL) is skipped because there are no orbitals to localize.
EIGS is skipped. Canonical eigenvalues not available during SCF optimization.
Polarization (KPOLAR, APOLAR) is skipped. Polarization involves numerical differentiation, which is very sensitive to the neglect of matrix elements.
Solvation (COSMO, SM5C, SM5.2) is very expensive. Currently, solvation is not able to take advantage of sparsity, so scales much like a full matrix calculation. It will work fine for small and medium sized systems but may be too expensive for large systems.
Standard optimizers (TRUSTE, TRUSTG, EF, TS, BFGS, DFP) are moderately expensive. Optimization routines are not fully linear in either CPU or memory but are still useful for molecules up to several thousand atoms. TRUSTE=LARGE greatly reduces the memory and CPU cost and so is usable up to about ten thousand atoms.
Second derivatives (LTRD, NEWTON, FORCE, LFORCE, HESSEI) are very expensive. Limited to several hundred atoms.
ANNEAL, IRC, CHN, PATH, reaction path, and reaction grid calculations will work but may be expensive or run into SCF convergence difficulties.
Mulliken charges (MULLIKEN) are quick but the storage of the Lowdin transformation (S-1/2) in the visualization file can be very large. Use NOVIS or VIS=MIN if it becomes a problem.
ESP charges (ESP) are expensive but are generally much less so that their full matrix counterparts for large systems. As with the Mulliken charges, storage of Lowdin transformation may be a problem for very large systems.
NBO's determination of the NLMO to MO transformation matrix (i.e. NBO=4) is skipped. All other parts of NBO are fully compatible with sparse.
Energy partitioning (ENPART) is linear in memory but weakly quadratic in CPU so is applicable to very large systems. The output file, however, may become very large but should not be prohibitive. The only real restriction is quadratic size of the storage in the visualization file. Using VIS=MIN to avoid quadratic storage if necessary.
Canonical orbitals (ALLVEC, VECTORS) are reduced. When either of these keywords are present, up to 10 canonical orbitals surrounding the homo-lumo gap are computed and printed (unless NOHL is also present).
SYBYL implies GRAPH, which is skipped. Also, eigenvalues will not be present in SYB file if NOHL keyword is present.
This section contains an alphabetical list of all keywords used with sparse matrix calculations.
|
Use DIIS during conjugate-gradient steps. |
|
|
Don’t store two electron integrals. |
|
|
Supplement the matrix form used in sparse PSOLVE with additional elements. |
|
|
Use Fletcher-Reeves version of conjugate gradient. |
|
|
Fully converge conjugate gradient at each SCF cycle. |
|
|
Use Gershgorin method to compute bounds on the Fock matrix eigenvalues. |
|
|
Generate initial guess based on Lewis dot structure analysis. |
|
|
Avoid computation of the HOMO-LUMO orbitals and gap. |
|
|
Do not use preconditioning during conjugate gradient. |
|
|
Set the convergence criteria for PSOLVE=CGDMS or QNDMS. |
|
|
Set level of output during LEWIS. |
|
|
Set the sparse matrix solver method. |
|
|
Set level of output during PSOLVE and sparse matrix operations. |
|
|
Use DIIS to improve convergence of the SCF. |
|
|
Perform sparse matrix calculation using the specified neglect threshold. |
|
|
Set level shift during CGDMS or QNDMS. |
[20] J. Chem. Phys.. 1997. 5569.
[21] J. Chem. Phys.. 1997. 425.
[22] J. Chem. Phys.. 1999. 1321.
[23] J. Chem. Phys.. 2003. 7651.
[24] Phys. Rev. B. 1998. 12704.
[25] Phys. Rev. B. 1995. 9455.
[26] J. Chem. Phys.. 1998. 3308.
[27] J. Chem. Phys.. 2000. 6035.
[28] Rev. Mod. Phys.. 1960. 335.
[29] Chem. Phys. Lett. 2002. 117.
|
Copyright © 1992-2013 Semichem, Inc. All rights reserved. |
|
|
 |
|
| Chapter 6. Keywords |  |
Chapter 8. CHN Methods |