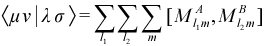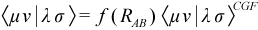| Chapter 3. Semiempirical Methods and Parameters | ||
|---|---|---|

|
Part I. AMPAC™ 10 User Guide |  |
| Chapter 3. Semiempirical Methods and Parameters | ||
|---|---|---|
|
|
Part I. AMPAC™ 10 User Guide | |
Table of Contents
MNDO, PM3, and AM1 are representatives of the NDDO (neglect of diatomic differential overlap) family of methods. As this model is applied to AM1 and PM3, there are seven basic parameters that must be considered for each atom. These are augmented with up to three (four in the special case of carbon) Gaussian corrections to adjust the core/core repulsion function (CRF). The two parameters with the largest effect are the one-center/one-electron energies, Uss and Upp. These represent the kinetic energy and core-electron attractive energy of single electrons in s- and p-orbitals. The next pair of parameters are adjustments to the two-center/one-electron resonance integral, β μν, and are termed β s and β p . These parameters are responsible for bonding interactions between atoms. Another approximation within AM1 is the use of Slater functions to describe spatial features of the atomic orbitals. Slater functions are used because they require fewer parameters and are more easily parameterized. The exponents of the s- and p-orbitals on each atom become parameters and are abbreviated respectively, ζ s and ζ p . It should be noted that ζ s and ζ p also affect βμν, as it is proportional to the overlap integral (Sμν), which is in turn calculated from the Slater exponents.
The CRF combines the Hamiltonian’s nuclear terms with core-electron repulsive and attractive effects. The α parameter scales the entire CRF up and down, providing a radial dimension to the atom’s core. The Gaussian correction functions added with AM1 operate on the CRF directly to correct it for specific defects. This type of correction can accommodate several chemical phenomena. One example is atoms that expand their octet when bonding, such as sulfur and phosphorous. Atoms with expanded octets are thought to use d-orbitals to allow back-donation of electrons for additional bonding. Although, AM1 has no explicit descriptions of d-orbitals, some of the effects d-orbitals have on bonding can be imitated by the Gaussian functions. Also, the single-ζ nature of the AM1 orbital basis set makes the method insensitive to some electronic factors and the Gaussians can mitigate these failures to some extent. For instance, semiempirical approximations tend to yield poor results for molecules where large charges are localized on atoms. The occupied orbitals of negatively charged species tend to expand in space due to electron repulsion. An analogous but opposite effect is noted for positively charged systems. These phenomena are accounted for in ab initio methods by using double- or triple-ζ basis sets or even additional special orbital functions (diffuse functions). This lack of flexibility in the AM1 orbital basis can be overcome to some degree by the Gaussian corrections. The use of Gaussians is an artificial correction made to the semiempirical treatment and should be avoided if possible. In some cases, however, Gaussians are beneficial and indeed necessary. Each Gaussian function consists of three parts: intensity (in eV), position (in Å) and width (in Å-2).
MNDO differs from AM1 and PM3 in that, for the lighter elements near the top of the Periodic Table, the following assumptions were made to save time:
Also, the MNDO method includes no Gaussian functions.
The following table shows which atom pairs are parameterized within the MINDO3 semiempirical model. A dot (•) indicates that calculations may be performed on an atom pair of the type shown.
Table 3.1. MINDO3 Atom Pair Parameters
| H | B | C | N | O | F | Si | P | S | Cl | |
|---|---|---|---|---|---|---|---|---|---|---|
| H | • | • | • | • | • | • | • | • | • | • |
| B | • | • | • | • | • | • | ||||
| C | • | • | • | • | • | • | • | • | • | • |
| N | • | • | • | • | • | • | • | • | ||
| O | • | • | • | • | • | • | • | • | ||
| F | • | • | • | • | • | • | • | |||
| Si | • | • | • | |||||||
| P | • | • | • | • | ||||||
| S | • | • | • | • | • | • | • | |||
| Cl | • | • | • | • | • | • | • |
Table 3.2, “Elemental Parameter Sets for all methods (except MINDO3)” lists the elements parameterized within the AM1, RM1, PM3, MNDO, MNDOC, MNDO/d, SAM1, SAM1D, and PM6 semiempirical methods. Again, a dot (•) indicates that the element has been parameterized. For MNDO/d, the diamond symbol (◊) indicates that the parameters are identical to the corresponding MNDO parameters, while a dot (•) indicates that the element has been re-parameterized for MNDO/d. For SAM1D, the diamond symbol (◊) indicates that the parameters are identical to the corresponding SAM1 parameters, while a dot (•) indicates that the SAM1 parameterization has been extended by adding d-orbitals.
Table 3.2. Elemental Parameter Sets for all methods (except MINDO3)
| Element | AM1 | RM1 | PM3 | MNDO | MNDO/d | MNDOC | SAM1 | SAM1D | PM6 | Element |
|---|---|---|---|---|---|---|---|---|---|---|
| H | • | • | • | • | ◊ | • | • | ◊ | • | H |
| He | • | He | ||||||||
| Li | • | • | ◊ | • | • | Li | ||||
| Be | • | • | ◊ | • | Be | |||||
| B | • | • | ◊ | • | B | |||||
| C | • | • | • | • | ◊ | • | • | ◊ | • | C |
| N | • | • | • | • | ◊ | • | • | ◊ | • | N |
| O | • | • | • | • | ◊ | • | • | ◊ | • | O |
| F | • | • | • | • | ◊ | • | ◊ | • | F | |
| Ne | • | Ne | ||||||||
| Na | • | • | Na | |||||||
| Mg | • | • | • | Mg | ||||||
| Al | • | • | • | • | • | Al | ||||
| Si | • | • | • | • | • | ◊ | • | Si | ||
| P | • | • | • | • | • | • | ◊ | • | P | |
| S | • | • | • | • | • | • | ◊ | • | S | |
| Cl | • | • | • | • | • | • | • | • | Cl | |
| Ar | • | Ar | ||||||||
| K | • | K | ||||||||
| Ca | • | Ca | ||||||||
| Sc | • | Sc | ||||||||
| Ti | • | Ti | ||||||||
| V | • | V | ||||||||
| Cr | • | Cr | ||||||||
| Fe | • | ◊ | • | Fe | ||||||
| Co | • | Co | ||||||||
| Ni | • | Ni | ||||||||
| Cu | • | ◊ | • | Cu | ||||||
| Zn | • | • | • | • | • | Zn | ||||
| Ga | • | • | Ga | |||||||
| Ge | • | • | • | • | Ge | |||||
| As | • | • | • | As | ||||||
| Se | • | • | • | Se | ||||||
| Br | • | • | • | • | • | • | • | • | Br | |
| Kr | • | Kr | ||||||||
| Rb | • | Rb | ||||||||
| Sr | • | Sr | ||||||||
| Y | • | Y | ||||||||
| Nb | • | Nb | ||||||||
| Mo | • | Mo | ||||||||
| Tc | • | Tc | ||||||||
| Ru | • | Ru | ||||||||
| Rh | • | Rh | ||||||||
| Pd | • | Pd | ||||||||
| Ag | • | Ag | ||||||||
| Cd | • | • | • | Cd | ||||||
| In | • | • | In | |||||||
| Sn | • | • | • | • | Sn | |||||
| Sb | • | • | • | Sb | ||||||
| Te | • | • | • | Te | ||||||
| I | • | • | • | • | • | • | ◊ | • | I | |
| Xe | • | Xe | ||||||||
| Cs | • | Cs | ||||||||
| Ba | • | Ba | ||||||||
| La | • | La | ||||||||
| Gd | • | Gd | ||||||||
| Lu | • | Lu | ||||||||
| Hf | • | Hf | ||||||||
| Ta | • | Ta | ||||||||
| W | • | W | ||||||||
| Re | • | Re | ||||||||
| Os | • | Os | ||||||||
| Ir | • | Ir | ||||||||
| Pt | • | Pt | ||||||||
| Au | • | Au | ||||||||
| Hg | • | • | • | • | • | Hg | ||||
| Tl | • | • | Tl | |||||||
| Pb | • | • | • | Pb | ||||||
| Bi | • | • | Bi |
SAM1 is the next step in the development process that produced MNDO and AM1. It uses a new theoretical basis for the underlying model and gives better results than previous models. SAM1 will also eventually allow the study of new elements (those that require d-orbitals) while retaining the traditional computational efficiency of semiempirical methods. The need for a new theoretical approach to semiempirical calculations is evident for two primary reasons. First, the known deficiencies of AM1 and MNDO in certain cases are resistant to removal by parameterization within the present theoretical model. The problems are inherent to the theoretical treatment and the only reasonable solution is a new method. Second, the present formalism for computing bicentric two-electron repulsion integrals (TERIs) is very cumbersome and difficult when applied to d-orbitals. At present AM1 and MNDO use a modified multipole expansion for computation of TERIs:
where M values are multipoles. The TERI values are computed by determining the magnitude of the electrostatic repulsions between the multipoles as described by Dewar, Sabelli, Klopman, and Ohno (DSKO method). While this expansion yields only 22 unique repulsion integrals for s- and p-orbitals, the number of integrals when d-functions are included is enormous. As has also been pointed out, integrals of this form are not rotationally invariant in the case of d-functions and the ME fails to converge to the appropriate one-center integral boundaries at short distances. A number of approaches have been attempted to “patch” the problems with DSKO repulsion integrals, but they either failed due to poor performance or reduce the flexibility of the semiempirical approximation to unacceptable levels.
The mode of calculation of TERIs is extremely important in semiempirical methods of the MNDO/AM1 family as other quantities depend on their values, e.g. the CRF (the repulsive force between core electrons) is simulated by use of an <ss|ss> integral. As noted above, the major difference between AM1 and MNDO is an additional modification of this function using a linear combination of Gaussian terms to correct for the inadequacies of the model. The core-electron attraction integrals (CEA, the attractive force between core electrons on one atom and nuclei on another) are also based on the TERI/ME formalism. With this in mind, it becomes clear that an alternative method of computing TERIs is a functional definition of a new semiempirical methodology. This new method of computing TERIs is the primary difference between SAM1 and the earlier NDDO semiempirical methods.
The approach chosen in the case of SAM1 is to utilize ab initio integrals, scaled using adjustable parameters so as to retain the implicit treatment of correlation effects, characteristic of the semiempirical methods. The form of the TERIs used is:
where <μν|λσ>CGF are ab initio integrals evaluated from contracted Gaussian basis functions fit to Slater type orbitals (STO-3G) using standard methods. The function f(RAB ) is a scaling function that features adjustable parameters, altered systematically to reproduce experimental data.
There are several significant advantages in the use of ab initio TERIs with a semiempirical procedure. First, the formulae are well defined and have been used for many years. Second, the gradients are easily computed from the Cartesian form of the Gaussian functions. This allows for the rapid and efficient construction of the Gaussians and their derivatives simultaneously. Third, formulae for d-orbitals already exist and these functions are bound by none of the limitations of the ME TERIs discussed earlier. It should be noted that the computation and manipulation of repulsion integrals is the most time-consuming (both CPU and I/O intensive) part of any ab initio calculation. The main reason for this is not the time required for a single integral calculation, but the large number of independent integrals which must be computed. In the SAM1 protocol, the NDDO approximation is still active, limiting the integrals which must be explicitly calculated to bicentric terms only, excluding three- and four-center terms from consideration. Because of the increased time spent on each integration, preliminary data shows that SAM1 is about two times slower than AM1. This is still well within acceptable limits when compared to ab initio treatments of similar accuracy.
Further details of this method are in preparation for publication.
The parameterization process allows assignment of specific values to quantities not directly available from experiment and difficult to estimate a priori accurately. The parameters may be based on either experimental data or the results of more complete HF calculations. The methods in AMPAC™ have traditionally depended on developing parameters based on experiment. This has significant advantages, first, in that any theoretical inadequacies of higher level ab initio methods are circumvented. Second, relaxation of the parameters in such a way as to reproduce experiment handles in one step a multitude of chemical factors not directly includable in the model.
The semiempirical approach used by Dewar et al. requires parameters to be derived for each type of atom in a molecule. Experimental data from a small group of representative molecules (which we refer to as the molecular basis set for parameterization, MBSP) is used to derive the parameter values. The items of data used are heats of formation (ΔHf, kcal/mol), ionization potentials (IP, eV), dipole moments (μ, Debyes), and molecular geometries (bond lengths, bond angles, and dihedral angles), all gas phase. Extensive experience has shown that the parameterization algorithm will eventually generate a set of parameter values that is extendible beyond the limits of those molecules in the MBSP. The derivation of the parameters proceeds by computing gradients of the parameters with respect to the experimental data. This generates an error function, SSQ, which is a weighted least-squares index quantifying the difference between calculation and experiment. A line search along this direction yields the optimum step size, and this information is used to update the Hessian (second derivatives) matrix which guides the search of the parameter hypersurface. The algorithm used in this regard is a modification of the Davidon-Fletcher-Powell procedure. New results are then calculated and compared with experiment. This series of steps is repeated iteratively until threshold values of gradient and SSQ are attained. The parameters are then considered to be optimized and testing may begin to determine their suitability for further calculations.
Several points must be noted about the parameterization process itself. It requires a systematic and tenacious approach and a large commitment of computational resources. The mathematical methodology will find some local minimum for the differences between experimental and calculated values (SSQ). This may not, however, be a minimum corresponding to sensible chemistry. The parameterization process requires constant attention to insure that the search algorithm (which tends to take occasional “sidetrips”) does not wander into an unprofitable region of the parameter hypersurface. Also, the minimum predicted by the program’s search algorithm is strongly dependent on the beginning values of the parameters. While projections for initial values can be made on the basis of the periodicity of chemical quantities (including semiempirical parameters), a reliable parameterization process requires two dimensional grid searches in the area of physical viability for the parameters. These searches help determine the best area of the parameter hypersurface from which to begin optimization. Grid searches are time-consuming and tedious, but necessary for proper refinement of the parameters. The molecules selected for the MBSP also have a great deal to do with the values that the parameters will finally assume. Every attempt is made to select species that represent important and useful chemistry without exceptionally biasing the results. In some cases, the MBSP must even be altered during the procedure to retain important aspects of the element’s chemistry and remove extraneous effects. The concept of using a small group of representative molecules for development of parameters to extend well beyond the MBSP has been tested and proven many times over, as exemplified by the large number of successful semiempirical parameter sets for several different methods produced by Dewar et al. over the past 30 years. It should also be noted that only non-radical (closed shell) and neutrally charged species are generally used in the MBSP. There are approximations within the method to specifically account for open shell species and parameters extendible to radicals and ions are reliably developed with this limitation.
Special considerations have been and will need to be made when dealing with transition metal systems and parameterization. First, the spin multiplicities and orbital mixing (CI) must be implemented and properly assigned as described above. Second, the data is limited in both quantity and quality. Both of these ideas lead to the idea that supporting calculations at the ab initio level will likely have to be done to support semiempirical calculations. They will likely fill “holes” in the data by providing correct spin multiplicities and geometries. Another point to note is the proper orbital description of the transition metal species will only be possible if parameterization is performed on molecules with the proper spin multiplicity pre-defined. This means that in addition to the traditional items used in parameterization (see above), spin multiplicity has been implicitly added to the list of data.
As pointed out both here and in the literature, the development of parameters is a difficult and tedious task. When done by the method here large amounts of computer time are required. This is especially true of the semi-ab initio protocol on which SAM1 is based. Parameterization requires a careful balance between acceptance of results from the search algorithm and intuition as to what is important and correct chemically: the HUMAN FACTOR.
Calculations involving transition metal elements is one of most important areas of chemistry that has been excluded from routine treatment by computational modeling. This is largely because methods that allow standard programs to implement high quality semiempirical theoretical approaches to systems containing these elements are only now being developed and are becoming available to the chemical community at large. SAM1 is one of the most successful of these new approaches. It should be emphasized at the outset of this discussion that only a limited amount of our accumulated wisdom and experience with organic and main group elements will be transferable to work on iron and other transition metals. It is a virtually new area using the Dewar-style semiempirical methodology. Previous experience has largely been with closed-shell systems (singlet spin state) and much more “regular” chemistry. This is certainly not the case with transition metals.
We began the parameterization of transition metals using a simple closed-shell approach as a way of exploring the problems that will be encountered. In this light, our preliminary efforts were very instructive and valuable. A number of obstacles (both expected and unexpected) were identified and have been overcome. Some of these are listed below.
development of new and larger CI algorithms and approaches
difficulty extrapolating initial values for the parameters from which to begin searches
ordering of the atomic orbitals
intelligent approach to CI for easier calculation and determination of the minimum level of CI required for a particular system
lack of high quality experimental data on spin multiplicity
The programmatic implementation of an “intelligent CI” is an important
addition to AMPAC in working with certain transition metal systems.
These include situations where the HOMO/LUMO MOs are degenerate to some degree, that should
lead to unpaired electrons with potentially high spin multiplicities. In this case extreme
caution must be exercised in choosing the correct OPEN(n,m) pattern or the reference wave function will
be a poor description and all subsequent calculations will be compromised. A segment of
program code has been added that will determine the degeneracies of the MOs near the HOMO/LUMO
boundary and suggest an OPEN pattern to use for this system to obtain a good reference wave
function. (The user can obviously override these default values if the need arises.) The
“natural” first guess orbitals from AMPAC are used to
commence the SCF procedure and to analyze the MOs. In this approach, the RHF wavefunction from
which the microstates are constructed for subsequent mixing is one where orbital occupancy and
degeneracy patterns are largely defined by the values of the parameters. Any method that is
developed ignoring this additional complication to more familiar semiempirical problems will
not be accurate or chemically meaningful. See Chapter 11, Configuration Interaction for
more information about configuration interaction in AMPAC.
RM1 is a reparameterization of AM1 and offers somewhat improved performance.[13] The theoretical form is exactly the same as AM1 and offers no improvement in underlying methodology. Careful re-optimization of the AM1 parameters using an improved dataset (both in quality and quantity) and better numerical methods resulted in a more accurate and precise method.
Table 3.3. Average Errors for Various Properties for H, C, N, O, P, S, F, Cl, Br and I (kcal/mol)
| Property | No. | AM1 | PM3 | PM5 | RM1 |
|---|---|---|---|---|---|
| ΔHf (kcal/mol) | 1480 | 11.15 | 7.98 | 6.03 | 5.77 |
| Dipole Moment (μ) | 127 | 0.37 | 0.38 | 0.50 | 0.34 |
| Ionization Potential (eV) | 232 | 0.60 | 0.55 | 0.48 | 0.45 |
| Interatomic Distances (Å) | 904 | 0.036 | 0.029 | 0.037 | 0.027 |
PM6 is a recently developed semiempirical method that follows in the tradition of other “Dewar-style” NDDO methods such as MNDO, AM1, PM3, SAM1, and PM5.[14] While slightly emphasizing biochemical systems in parameterization, PM6 is a general purpose model of good quality.
The “PM” of the method’s name stands for “parameterization method”, and this is the primary difference between PM6 and previous NDDO methods. Stewart set out to evaluate and systematize the accreted approximations and changes to recent semiempirical method development. Some of the most important aspects of PM6 that differ from previous work include:
Careful examination and pruning of the experimental data to ensure high quality and consistency
Use of ab initio and DFT results where experimental data is lacking
Use of “rules” that allow different experimental data (such as dimerization energies) to be included in parameterization
The addition of a large number of pairwise interaction terms that adjust the core repulsion function
One-center two-electron integrals for s- and p-orbitals are computed differently for main group and transition elements
Special corrections for nitrogen pyramidalization and carbon triple bonds (see next section)
The performance of PM6 is better that that for AM1, PM3, RM1 or PM5, as shown on the table below. Similar results are obtained for ionization potentials, dipole moments and geometries
Table 3.4. Average unsigned errors in ΔHf for various sets of elements (kcal/mol)
| Set of Elements | No. | PM6 | RM1 | PM5 | PM3 | AM1 |
|---|---|---|---|---|---|---|
| H, C, N, O | 1157 | 4.64 | 4.89 | 5.60 | 5.65 | 9.41 |
| H, C, N, O, F, P, S, Cl, Br, I | 1774 | 5.05 | 6.57 | 6.75 | 8.05 | 12.57 |
| All Main Group | 3188 | 6.16 | - | 15.27 | 17.76 | 22.34 |
| All 70 PM6 Elements | 4492 | 8.01 | - | - | - | - |
Stewart introduced two special Molecular Mechanics (MM) style corrections into his implementation of PM6 to correct the chemistry of specific systems. AMPAC has modified these corrections and added a third correction for H2. It is important to discuss each of these additions. As such, AMPAC will not exactly reproduce MOPAC® results for these cases.
A correction term was added to compensate for an overestimation of the degree of pyramidalization in secondary and tertiary amines. (Primary amines did not require any correction.) A MM-style term was added to favor a more planar configuration. Stewart’s original formulation had to be modified because the bonding analysis he used to determine when to apply this term did not evolve to match changes in the geometry and so can give rise to strange inconsistancies. Simply updating the bonding at each new geometry was not enough, however, because this led to discontinuities in the potential energy surface. (Discontinuities can stall optimizations, produced fall minima, or cause other problems.) As such, AMPAC modified Stewart’s original formulation by adding smoothing functions to allow a gradual transition from bonded to non-bonded to eliminate these discontinuties.
The carbon-carbon triple bond is very important in chemistry and so efforts were taken to insure that it was modeled correctly. Stewart added a 12 kcal/mole MM-style correction term for carbon-carbon triple bonds that is in addition to his special modifications to the core-core interaction terms. (This MM correction, however, is not discussed in his paper.) As with the amine correction, this term in Stewart’s formulation was added inconsistently because the bonding information did not change with the geometry. It was not, however, possible to correct this using smoothing fuctions without disturbing double bonds or introducting spurious minima. As such, AMPAC simply mimics Stewart’s original coding.
An additional change was made to Stewart’s original PM6 to improve accuracy of H2. A polynomial is used in the core-core interaction to correct the behavior of H-H bonds at short distances to match known heat of formation, bond length, and frequency data. Longer H-H distances, such as in water, are unaffected by this change.
Use NC-PM6 with PM6 to disable all three of these correction terms.
|
Copyright © 1992-2013 Semichem, Inc. All rights reserved. |
|
|
 |
|
| Chapter 2. Special Features of AMPAC |  |
Chapter 4. Computational Procedures |